Surgery, Gastroenterology and Oncology

|
|
Budd-Chiari syndrome (BCS) represents a rare medical entity which has an estimated incidence of 0.1 to 10 people per million every year. It is defined by the obstruction of the flow in the inferior vena cava or the hepatic veins. Various classifications have been proposed. So, it can be acute or chronic and primary or secondary. The chronic form is more frequent and is characterized by signs of portal hypertension. Liver transplantation is the ultimate therapeutic management of Budd-Chiari syndrome. Primary BCS is mostly a result of hematological disorders and hypercoagulable conditions. Secondary BCS appears due to invasion or extrinsic pressure of the veins from various reasons, including hepatocellular carcinoma (HCC), liver abscesses and cysts. We presented a rare case of a young lady with Budd Chiari syndrome and IVC thrombosis who were transplanted with hepatic left lobe from her sister.
INTRODUCTION
Budd-Chiari syndrome (BCS) is a rare disease involving obstruction of the hepatic venous outflow at various levels from the small hepatic vein to the suprahepatic lesion of the inferior vena cava (IVC) (1,2). This hepatic venous obstruction increases hepatic sinusoidal pressure, followed by portal hyper-
tension, which causes liver injury and finally liver cirrhosis. When various treatments for patients with BCS have failed, and liver cirrhosis is decompensated, liver transplantation remains the only curative option (2,3). In deceased donor liver transplantation (DDLT) for patients with BCS, suprahepatic caval resection and replacement IVC is the standard procedure. However, due to the absence of an IVC in the liver graft in living donor liver transplantation (LDLT), there are technical difficulties in the implantation procedure in terms of the construction of hepatic venous outflow (3). In adult-to-adult LDLT, right-lobe graft and left-lobe graft transplants are mainly used. However, this sometimes involves difficulties with limited graft selection, particularly in terms of insufficient graft volume for recipients and a small residual liver volume in the donor.
CASE REPORT
A 35 years old lady who has no children, with a medical history for the use of combined oral contraceptives from 16 years old.
She had been diagnosed with Budd-Chiari syndrome when she was referred with pilonidal abcess, at 22 years old, in December 2009. Her clinical examination revealed lower limb thrombosis with eritematous skin in inferior limb, leg heaviness, pain, swelling and cramps. Also, the presented with dilatation of superficial abdominal vessels and mild ascites.
Laboratory testing for autoimmune hepatitis was negative, as were serological markers for hepatitis C and B viruses. The patient also denied previous alcohol abuse. No thrombophilia was diagnosed, despite extensive hematological investigation.
Biological results: Hb 10,6 g/dL, PLT 144000/mmc, WBC 10800/mmc,INR=1,08, ALT -54 UI/mL, AST -98 UI/mL, TBIL- 0,91 mg/dL, GLC- 77 mg/dL, AFP=7 UI/mL, GGT-174 mg/dl; FALC-102U/L.
Unomogenous hepatomegaly with sg I hypertrophy, an axial diameter of 50 mm, with no intrahepatic nodules identified. Suprahepatic veins filiforme with complete obstruction of left SHV.
Multiple abnormalities of subdiafragmatic veins, with the ectasy of the azygos system, inferior costal and lombar veins; aberrant drainage of the hemiazygos vein in left renal vein, ascites, moderate splenomegaly.
In december 2009 she started anticoagulant therapy - acenocumarol (sintrom) with good response almost 8 years but from september 2017 her clinical condition slowly progressively worsened. The MRI was performed and it described liver with increased dimensions the cranio-caudal diameter of the right lobe 17 cm, hypertrophy of the caudal lobe in progression with the axial diameter of 54 mm and cranio-caudal 60 mm, collateral circulation routes in progression compare to the previous exam, moderate splenomegaly and progressive ascities.
In the same time, we repeated the complete hematological investigation: JAK2V617F mutation- negative; CALR (calreticuline) of the 9 axon – negative; MPL mutation (trombopoetine receptor) W515L/K/A and S505N – negative; ANA, ASMA,AMA, c ANCA, p ANCA, Ac anti Sm, Ac anti Ro , CIC, C3,C4, Ac anti SLA, Ac anti dc, Ac anti ADN double catene- negative;
The standard blood count showed cholestasis and minimu anemia with Hb – 10,8 g/dl, WBC - 8000, PLT-275000, ALT-15 u/ml, AST-25 u/ML, Tbil- 0,9 mg/dl, GGT-192 mg/dl; FALC-98U/L.
The evaluation of thrombophylic screening presented normal value of Ac anti fosfolipidic DRWT, Mixon LA, Ac anti cardiolipin Ig M and IgG, Ac anti B2glicotrotein IgM and IgG, V Leiden factor, ATIII, PC, PS, TPZ, INR - normal value.
The only continuous modified parameter was VIII factor - 193,3% (68-133%).
A long period of VIII factor > 150% represent a 5 time risqué factor of venous thrombosis; this factor is a acute phase proteine and can be ingrease temporary in many clinical situations: infections, stress, malignicies, hepatic or renal disorders, after surgical stress.
In October 2017-it was performed cutaneous biopsy: nodular erythema vs peripheral vasculitis and the histopathological aspect was compatible with the diagnosis of lymphocytic vasculitis of small/medium vessels. The decision was to start cortisteroid therapy, but after three months it was found progressive disease with voluminous ascities-and it was stopped cortico-therapy.
In february 2018 - her family decided to complete the evaluation in Frankfurt where medical team decided to evaluate the patient for TIPS, the choise treatment of severe portal hypertension complications. The cavography described complete thrombosis of IVC at 2-3 cm before right atrium, with a P= 24 mmHg in the cranial part of IVC and in SHV. Because TIPS was unfortunately impossible, the DIPS procedure was performed.
The Direct Intrahepatic Portocaval Shunt (DIPS) is a modified transjugular intrahepatic portosystemic shunt (TIPS) procedure, in which a stent is placed across the inferior vena cava directly into the portal vein under intravascular ultrasound guidance (fig. 1).
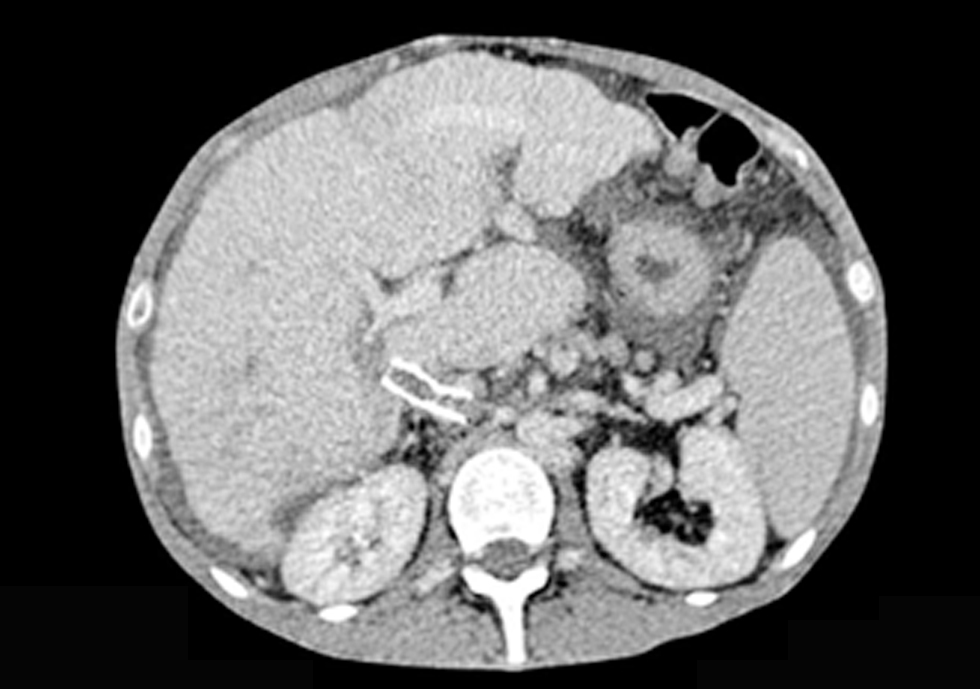
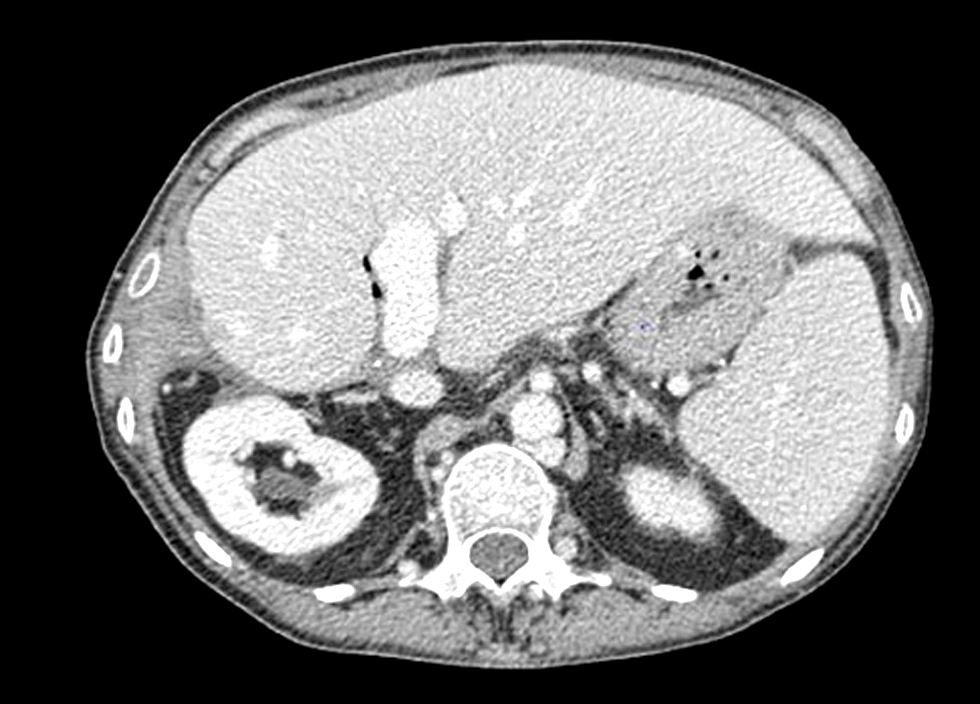
Our patient received continue theraphy with dabigratan etexilat (Pradaxa) 9 month and she performed CT scan every 3 month. In june 2018 CT scan evaluation described moderate ascities due to DIPS thrombosis (figs. 2, 3, 4).
Figure 1 - Direct Intrahepatic Portocaval Shunt (February 2018)

Figure 2 - CT scan 1.5 mm arterial - Direct Intrahepatic Portocaval Shunt thrombosis (June 2018)
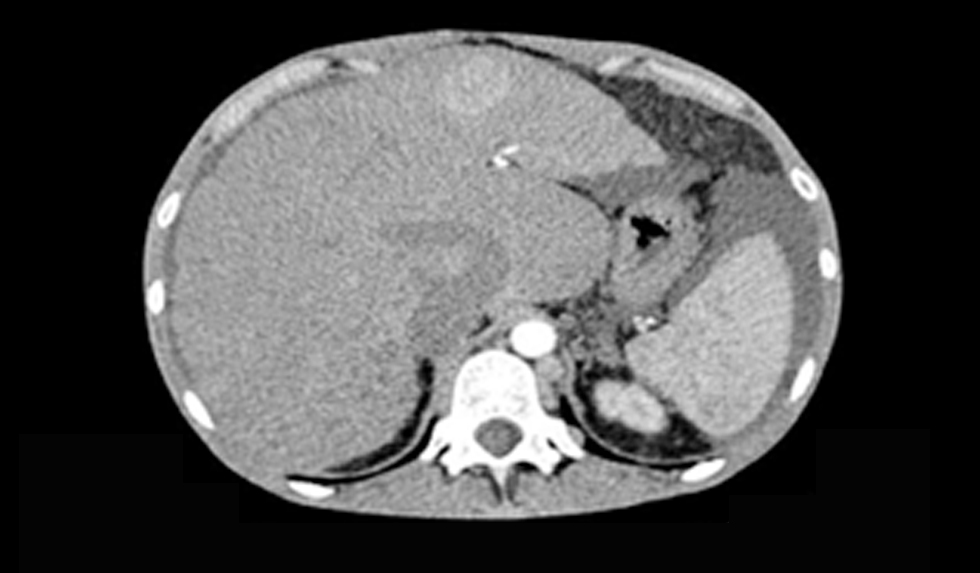
Figure 3 - CT scan 1.5 mm coronal - Direct Intrahepatic Portocaval Shunt thrombosis (June 2018)

Figure 4 - CT scan 1.5 mm portal - Direct Intrahepatic Portocaval Shunt thrombosis (June 2018)
There was no technical possibilities to re-open the DIPS. In this situation, our pacient incouraged by her family, decided to move to Viena University Hospital where the medical team managed to implant a device used to shunt ascites to the superior vena cava in patients with refractory ascites, a peritoneovenous shunt called Denver shunt.
The proximal end is located in the peritoneal cavity and the distal end in the superior vena cava via the internal jugular or subclavian route, with a subcutaneous course in the anterior chest wall.
It has a one-way valve and a compressible chamber, which must be compressed several times a day to ensure proper flow.
Figure 5 - Denver catheter

Contraindications to the procedure include: end-stage renal failure requiring dialysis, sepsis, uncorrectable coagulopathy, morbid obesity, multiple septs on the peritoneal cavity due to previous infection or surgery.
Recognized complications include: shunt occlusion, peritoneal infection, ascitic leak, bleeding, pulmonary edema, disseminated intravascular coagulation, pneumothorax, pneumoperitoneum.
After the insertion, she pumped 40 min /daily, 10 min 4 time in the day. After 2 years, it was need only 5 min per day. Our pacient had a acceptable quality of life from nov.2018 to july 2021, with regular examination every 3 month in Viena Hospital, but in 2021, the ascities start to increase, she used the pump until 1h: 30 min per day (figs. 5, 6).
Figure 6 - Clinical aspect before and after Denver implantation
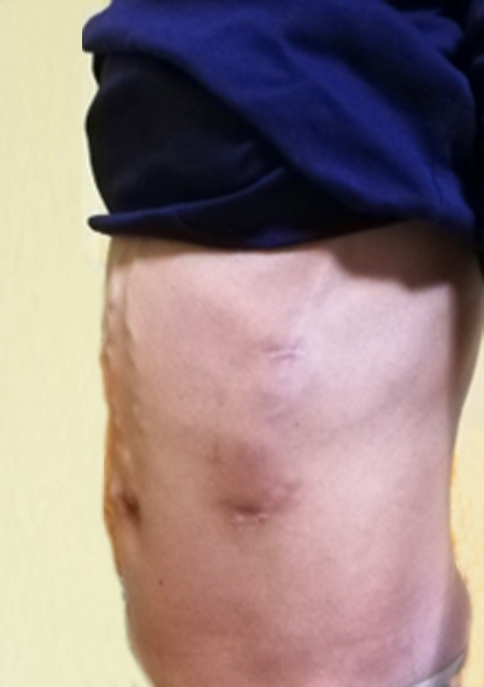
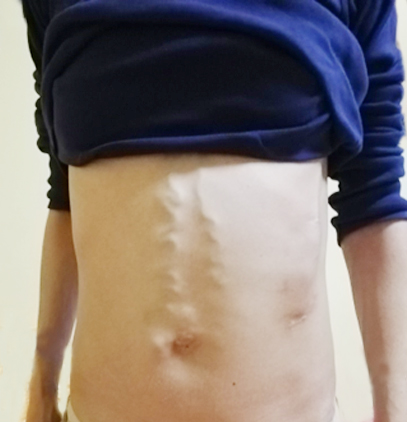
In July 2021 the medical team from Viena University Hospital advised her to accelerate the investigations for liver transplant in her country in the next 6 month.
We decided to start the specific protocol for LT with standard blood tests, superior endoscopy: grade I esofageal varices and cardiologic examination. She had malnutrition with BMI-18 and progressive ascites which needs paracentesis.
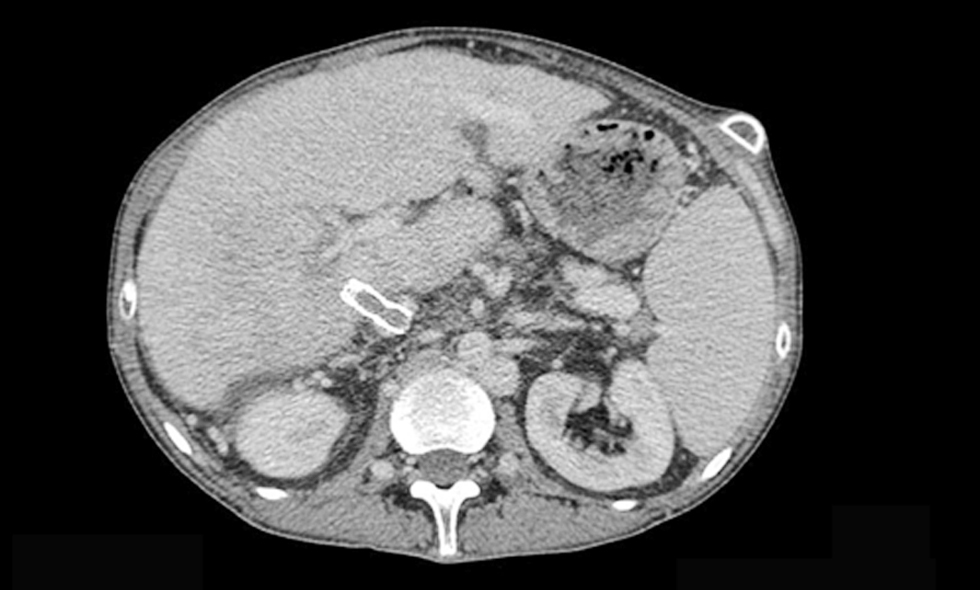
In October 2021 her CT scan revealed a cirotic liver with important portal hypertension (fig. 7).
In the same time, we started to investigate her husband as potential liver donor. Unfortunately, his hepatic volumetry on left lobe was 394 cmc - 27,3% from a total volume: 1441 cmc, so a GWRW less than 0.8.
Figure 7 - CT scan - cirrhotic liver with important portal hypertension (October 2021)
Her sister was the right potential donor with a total volume: 1276 cmc, right hepatic lobe: 800 cmc - 62% and left hepatic lobe: 476 cmc - 38 %.
In November 2021, SARSCOV 2 infection in both donor and receptor determinated to delay the surgical intervention until december 2021 when it was performed living related liver transplantation with left lobe for decompensated hepatic cirhosis due to Budd-Chiari syndrome, IVC thrombosis and refractory ascities.
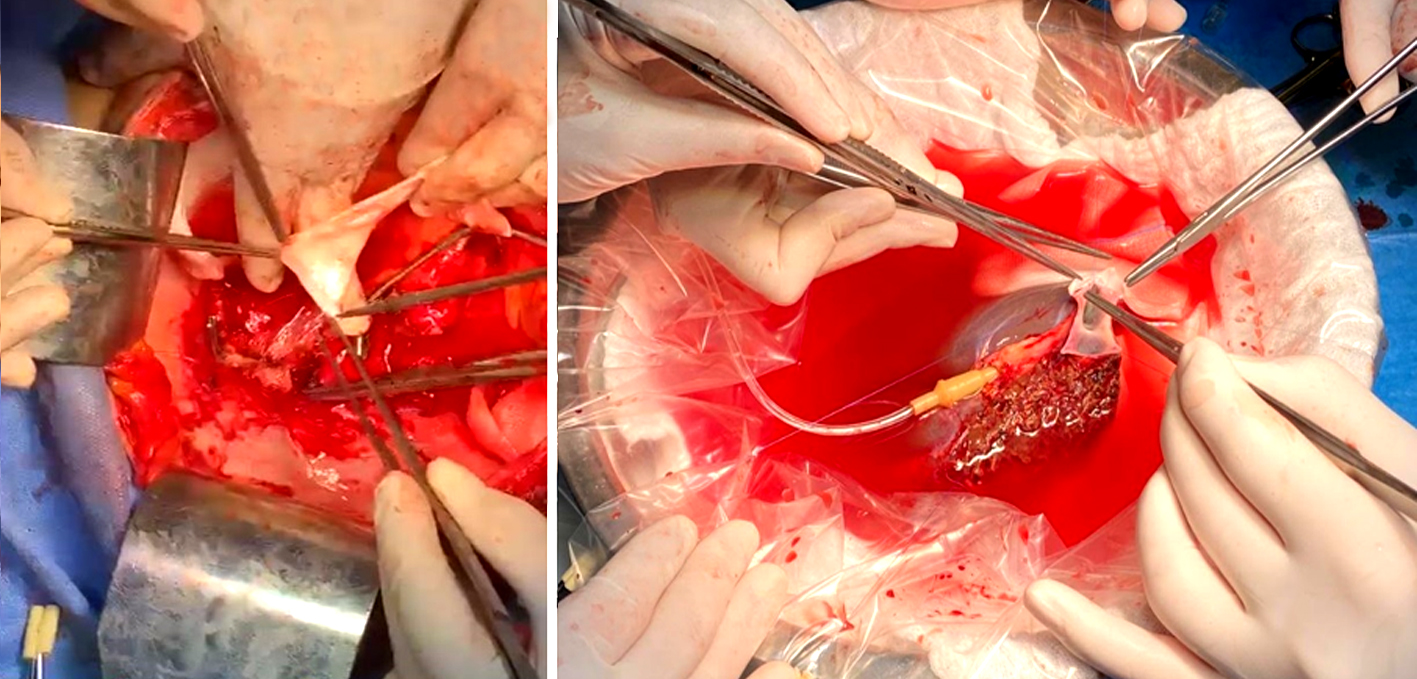
The orthotopic liver transplant with left hemiliver living donor had the following anastomoses:
-
Donor middle and left hepatic veins common trunk (Venoplasty) - recipient inferior vena cava end-to-side anastomosis (interposition of a cadaveric venous patch);
-
Donor portal vein - recipient left portal vein end-to-end anastomosis;
-
Recipient common hepatic artery - donor left hepatic artery end-to-end (microscope used for suture);
-
Cholangio-jejunal Roux-en-Y anastomosis (trans jejunal stenting with transjejunal exteriorization).
The imunosupresion was done with Basiliximab induction follow by Tacrolimus and Micofenolat Mofetil and CMV prophilaxy with valganciclovir (fig. 8).
Figure 8 - Intraoperative aspect
In postoperative ATI period several complications appeared: ascities, bilateral pleuresia which needed drainage, major coagulation disorders and secondary bleeding without endoscopic sourses of melena correctable with MER and coagulation factors AT III with a good evolution, no other hemorage episodes and no thrombosis. At this moment we decided to suprime Denver catheter.
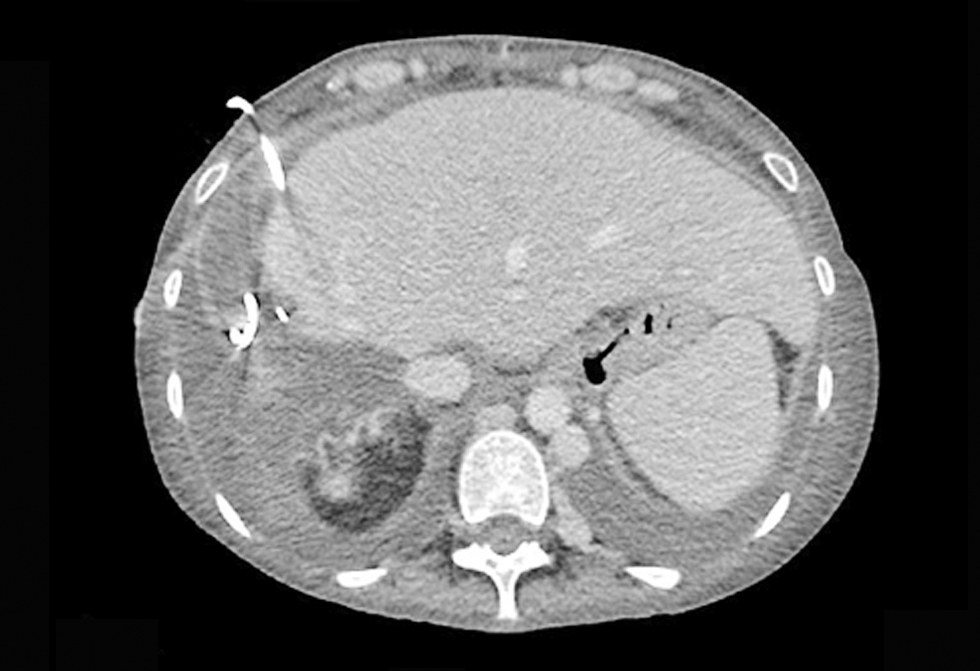
In January 2022 our patient started to present fever and leukocytosis. The ultrasonogrophy exam and CT scan described an intraabdominal collection and a percutaneous drenage almost 1600 ml and 600 ml/day was done. The complex antibiotherapy due to a multidisciplinary evaluation was necessary and because of hematologic disorders with agranulocytosis secondary to mycofenolat mofetil and valganciclovir treatment, steroid therapy was initiated with dexametazone (fig. 9).
Figure 9 - CT scan intraabdominal collection – percutaneous drenage (January 2022)
The progressive hematologic disorders needed multiple plasma exchange procedures and the biological picture revealed WBC-2630; Hb-7,1; Ht-21,3%; PLT-33000; INR-2,02; fibrinogen-199; ALT-8; AST-17; chol total-48; Tbil-1,1; creat-2,28; uree-74,9. For 10-14 days heparinotherapy and ATIII concentrate i.v with monitoring AT III factor was used, with continous renal replasment with additional filters for cytokines removal Cytosorbe and Oxiris.
In February 2022 – she was transferred in surgical department with ameliorate parameters. In march 2022 bone biopsy was performed with very rich marrow, intensely huperplastic on the megakaryocytic series, hyperplastic on the red series, dysplastic on all series, intensely on the megakaryocytic series and cortico-steroid therapy was started.
In may 2022 she was discharge with anticoagulant therapy, corticosteroids, IPP, imunosupression with advagraf.
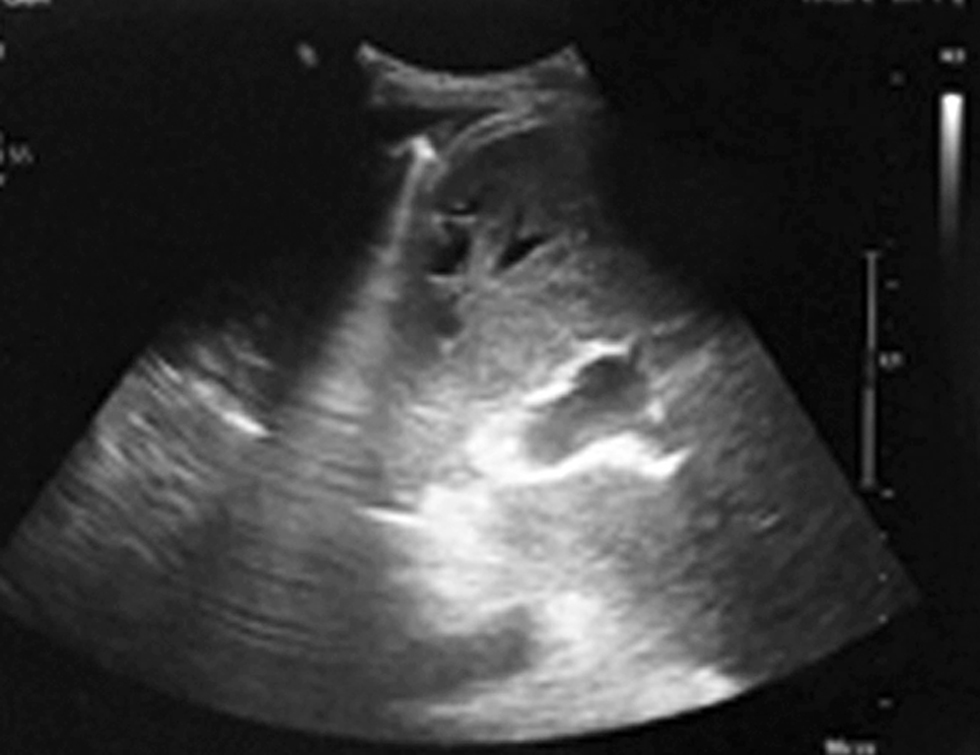
The ultrasound examination in September 2022 was with normal hepatic parenchima, patent vascular anastomosis and without collection or ascities (fig. 10).
Figure 10 - Ultrasound examination - no more perihepatic collection (September 2022)
The patient’s condition is good at her last follow-up, 1 year after transplantation (fig. 11).
Figure 11 - CT scan screening evaluation –normal aspect after LDLT (September 2022)
DISCUSSION
Budd-Chiari syndrome (BCS) is a rare disease resulting from obstruction of the hepatic venous outflow tract which leads to deterioration of liver function. Sometimes it can be caused of acute liver failure, that requires an emergency liver transplantation (6). The most common cause in Western countries is hyper-coagulability status - myeloproliferative disorder, the most commonly polycythemia vera. In Asian countries the most common cause is membranous obstruction (partial or complete) of the inferior vena cava (MOVC) and/or the hepatic veins. The treatment requires: anticoagulation, angioplasty/stenting - TIPS1, surgical shunt and liver trasplantation. Thus, BCS requires a multidisciplinary approch including: haematology, hepatology, interventional radiology and liver surgery.
For patients with BCS, endovascular treatments, such as angioplasty, stenting, and local thrombolysis, are first conducted. In patients with liver cirrhosis and portal hypertension, a transjugular intrahepatic portosystemic shunt or surgical venous shunt should be considered to decrease the portosystemic gradient (1). Additionally, anticoagulation therapy is recommended for all patients with BCS to prevent the progression of thrombosis. Although there are several treatments for BCS, including anticoagulation and endovascular treatment, liver transplantation is the only curative treatment modality for decompensated patients with end-stage liver disease or acute deterioration.
CONCLUSION
LT is performed in 10% to 20% of BCS patients.
This is a challenging indication for LDLT due to a combination of massive liver and presents thrombosis of IVC in LDLT, in which the donor’s IVC cannot be used, the retrohepatic IVC dissection performed during the piggyback technique and the venous outflow reconstruction are particularly problematic. Despite the complexity of cases, most studies describe successful outcomes after LDLT.
In our case, the particularity is the long history of the disease, with bridging therapy- DIPS and Denver shunt and co-association with limfocitar vasculitis associated with the difficulty of venous outflow reconstruction and the complexity of post-operative evolution. Four BCS patients were transplantated in Fundeni Clinical Institute, only this patient with left lobe graft.
Conflict of interest
All author declare that they have no conflict of interest.
Ethical Statement
All procedures performed were in accordance with the ethical standards of the 1964 Helsinki Declaration and its later amendments.
REFERENCES
1. Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med. 2004;350:578–585.
2. Valla DC. Primary Budd–Chiari syndrome. J Hepatol. 2009;50: 195–203.
3. Akamatsu N, Sugawara Y, Kokudo N. Budd–Chiari syndrome and liver transplantation. Intractable Rare Dis Res. 2015;4:24–32.
4. Sugawara Y, Makuuchi M, Takayama T, Mizuta K, Kawarasaki H, Imamura H, et al. Liver transplantation using a right lateral sector graft from a living donor to her granddaughter. Hepatogastro-enterology. 2001;48:261–263.
5. Valla DC. Hepatic venous outflow tract obstruction etiopathogenesis: Asia versus the West. J Gastroenterol Hepatol. 2004;19:S204–S211.
6. Parekh J, Matei VM, Canas-Coto A, Friedman D, Lee WM, Acute Liver Failure Study Group. Budd-chiari syndrome causing acute liver failure: A multicenter case series. Liver Transpl. 2017;23(2): 135-142.
Full Text Sources:
Abstract:
Views: 2828

For Authors
Journal Subscriptions

Dec 2025
Supplements
Instructions for authors
Online submission
Contact
e-ISSN: 2601 - 1700 (online)
ISSN-L: 2559 - 723X
Journal Abbreviation: Surg. Gastroenterol. Oncol.
Surgery, Gastroenterology and Oncology (SGO) is indexed in:
- SCOPUS
- EBSCO
- DOI/Crossref
- Google Scholar
- SCImago
- Harvard Library
- Open Academic Journals Index (OAJI)
Surgery, Gastroenterology and Oncology (SGO) is an open-access, peer-reviewed online journal published by Celsius Publishing House. The journal allows readers to read, download, copy, distribute, print, search, or link to the full text of its articles.
Time to first editorial decision: 25 days
Rejection rate: 61%
CiteScore: 0.2

Meetings and Courses in 2025
Meetings and Courses in 2024
Meetings and Courses in 2023
Meetings and Courses in 2022
Meetings and Courses in 2021
Meetings and Courses in 2020
Meetings and Courses in 2019
Verona expert meeting 2019

Surgery, Gastroenterology and Oncology applies the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits readers to copy and redistribute the material in any medium or format, remix, adapt, build upon the published works non-commercially, and license the derivative works on different terms, provided the original material is properly cited and the use is non-commercial. Please see: https://creativecommons.org/licenses/by-nc/4.0/
Publisher’s Note:
The opinions, statements, and data contained in article are solely those of the authors and not of Surgery, Gastroenterology and Oncology journal or the editors. Publisher and the editors disclaim responsibility for any damage resulting from any ideas, instructions, methods, or products referred to in the content.